雷特綜合征是一種罕見的神經發育障礙性疾病,屬于X染色體連鎖顯性遺傳病,主要影響女性。該病以發育倒退、手部刻板動作、語言及運動功能喪失為典型特征,由MECP2基因突變引起。

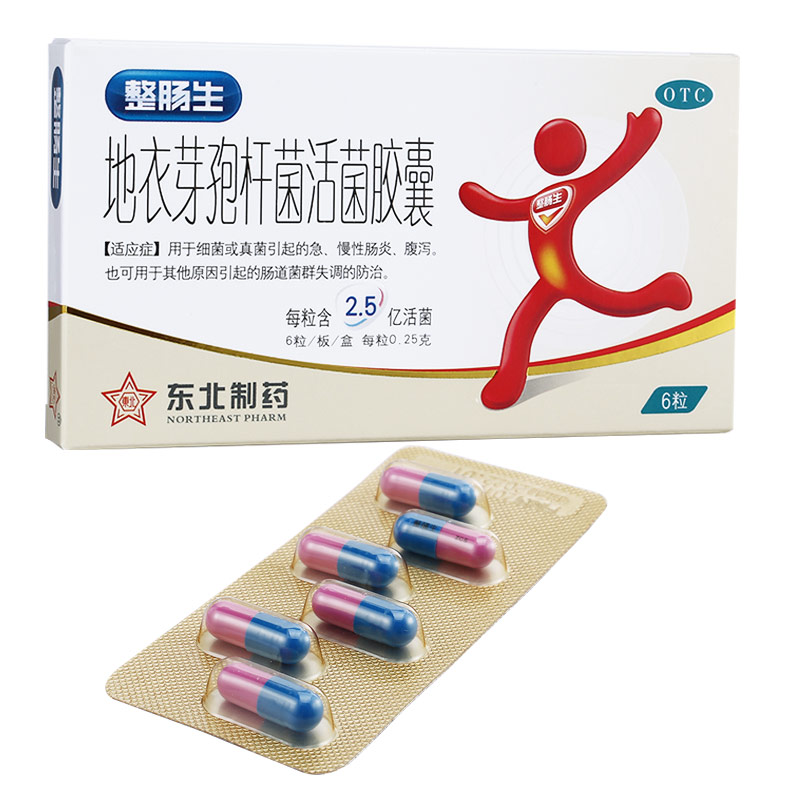
丙戊酸鈉
生產廠家:湖南省湘中制藥有限公司
功能主治:本藥為原發性大發作和失神小發作的首選藥,對部分性發作(簡單部分性和復雜部分性及部分性發作繼發大發作)療效不佳。對嬰兒良性肌陣攣癲癇、嬰兒痙攣有一定療效,對肌陣攣性失神發作需加用乙琥胺或其他抗癲癇藥才有效。對難治性癲癇可以試用。本藥除用于抗癲癇外,還可用于治療熱性驚厥、運動障礙、舞蹈癥、卟啉癥、精神分裂癥、帶狀皰疹引發的疼痛、腎上腺功能紊亂,以及預防酒精戒斷綜合征。
用法用量:1.成人常用量:每日按體重15mg/kg或每日600~1200mg分次2~3次服。開始時按5~10 mg/kg,一周后遞增,至能控制發作為止。當每日用量超過2500mg時應分次服用,以減少胃腸刺激。每日最大量為按體重不超過30 mg/kg、或每日1.8~2.4g。2.小兒常用量:按體重計與成人相同,也可每日20~30mg/kg,分2~3次服用或每日15 mg/kg,按需每隔一周增加5~10 mg/kg,至有效或不能耐受為止。
立即購買
雷特綜合征的臨床表現分為四個階段。早期階段6-18個月表現為發育停滯,患兒逐漸喪失已獲得的語言和手部功能。快速倒退期1-4歲出現典型的手部重復搓扭動作、呼吸節律異常及社交退縮。假性穩定期2-10歲運動障礙加重,可能出現脊柱側彎和癲癇發作。晚期運動惡化期10歲以上以進行性運動功能退化為主,多數患者需要輪椅輔助。
診斷需結合臨床特征和基因檢測。MECP2基因檢測是確診依據,約95%典型病例可檢出突變。鑒別診斷需排除自閉癥譜系障礙、腦性癱瘓等其他神經發育疾病。
治療采取多學科綜合管理。康復訓練重點改善運動功能和日常生活能力,作業療法可緩解手部刻板動作。抗癲癇藥物如丙戊酸鈉、拉莫三嗪用于控制發作。脊柱側彎超過40度需考慮矯形手術。營養支持對存在吞咽困難的患者尤為重要。
該病預后與突變類型相關。典型病例平均生存期約50歲,主要死因包括呼吸系統并發癥和癲癇持續狀態。定期監測心肺功能、骨密度及胃腸狀況可改善生活質量。目前針對基因治療的臨床試驗正在進行中。